How ALS Progresses on Genetic and Cellular Level Revealed by High-Res Spinal Cord Study

Precise experiments have revealed for the first time how Lou Gehrig’s disease, or amyotrophic lateral sclerosis (ALS), progresses on a genetic and cellular level. The work opens new avenues for developing potential treatments for the disease, which affects around 450,000 people worldwide.
“This is a whole new view of the disease,” says Tarmo Äijö, a research scientist at the Center for Computational Biology at the Flatiron Institute in New York City. “The hope is to figure out what is causing ALS and, in the future, come up with therapies.”
Äijö spearheaded the research alongside Silas Maniatis of the New York Genome Center and Sanja Vickovic of the New York Genome Center, the Broad Institute of MIT and Harvard, and the KTH Royal Institute of Technology in Stockholm. The researchers report their results in the April 5 issue of Science.
ALS is a neurodegenerative disorder that kills the neurons in the brain and spinal cord responsible for voluntary muscle movement. The loss of those neurons can cause muscle weakness, paralysis and eventually death. Baseball legend Lou Gehrig and astrophysicist Stephen Hawking both had ALS. (In 2014, the ALS Association’s Ice Bucket Challenge raised funds and increased public awareness of the disorder; the association supported the new study.)
The cause of ALS isn’t known, though previous work identified genetic factors that play a role. Scientists can track whether a given gene is activated, or ‘expressed,’ by looking for the gene’s messenger RNA molecule, or mRNA, which acts as a blueprint for building proteins that carry out various functions.
Experimental limitations have stymied efforts to better understand gene expression changes over the course of the disease. Existing techniques struggle to take large numbers of measurements with the high spatial resolution needed to track the progression of ALS at the smallest scales.
For the new study, the researchers employed an innovative approach recently developed by Vickovic and colleagues. The method involves placing thin tissue samples onto glass slides, with each slide covered by 1,007 tiny spots. Each spot is roughly the size of a dust mite and contains molecules that capture mRNA. The captured mRNA is then copied, and a unique genetic sequence or ‘bar code’ that corresponds to that particular spot on the slide is encoded in the copy. These bar codes enable the researchers to analyze all the spots at once and still link each mRNA molecule to its original spot.
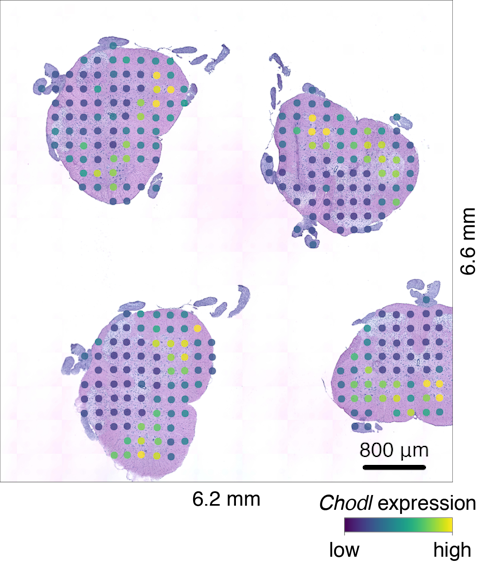
The researchers used the slides to collect more than 76,000 gene expression measurements from 1,200 spinal cord tissue sections of mice. Additionally, the researchers took more than 60,000 gene expression measurements from 80 postmortem spinal cord tissue sections donated by patients with ALS.
The mouse samples included individuals at many stages in the disease’s progression, enabling the researchers to track gene expression over time. In the human samples, the researchers compared the expression of genes collected from either end of the spine. ALS symptoms typically appear first in one part of the body, eventually spreading and causing widespread paralysis. By studying two spinal cord locations from each patient, the researchers were able to gain a view of the processes involved in the spread of ALS pathology.
The team now had tens of thousands of measurements, but analyzing the data proved challenging. Many spots yielded little to no genetic information, and it was unclear how the researchers could extract interpretable information from the vast amount of data they had generated. The computational challenges involved piqued Äijö’s interest when Maniatis presented preliminary work at a seminar hosted at the Flatiron Institute.
Äijö analyzed the data alongside Flatiron Institute colleagues Richard Bonneau, who serves as group leader for systems biology, and senior software engineer Aaron Watters. Their analysis provided gene expression information across three scales: individual spots, neighboring spots and whole anatomical regions.
This work provided numerous insights into the progression of ALS. The data suggest that specialized cells called microglia responsible for removing damaged neurons exhibit dysfunction well before the onset of ALS symptoms. Furthermore, the researchers found that two genes expressed by microglial cells, TREM2 and TYROBP, are expressed at higher levels in particular spinal cord regions of mice with ALS symptoms. Previous studies had suggested a link between the two genes and ALS, but using their new approach, the researchers were able to detect these changes far earlier, and to describe in unprecedented spatial detail how the behavior of microglia changes. The new work also describes disease-related changes in many cell types and in the ways cells communicate with one another.
Further research is needed to determine whether any of the findings indicate a potential root cause of ALS, or if they are merely part of the body’s response to the disease. The vast dataset generated in this study may help researchers to develop therapeutic interventions that thwart the mechanisms responsible for ALS, or to identify diagnostic markers that could allow detection of the disease before symptoms appear.
According to Äijö, the new work also highlights the importance of collaboration in studying ALS.
“We all brought different skills to the table, and this couldn’t happen without all of us,” he says. “The people in Stockholm have the techniques to do these measurements, and then the people at the New York Genome Center have the understanding and the knowledge of ALS, and then we at the Flatiron Institute have the computational expertise. This was really an extraordinary collaboration.”
An interactive data exploration portal is publicly available at http://als-st.nygenome.org/.
Information for Press
For more information, please contact [email protected].


