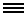

Image Credit: Guoping Feng Laboratory/MIT
Adult mice with a non-functioning copy of the Shank3 gene are at greater risk of autism-like symptoms. Neurons with the mutation have fewer synapses (middle) than typical mice (left), work by Feng and colleagues shows. This synapse deficit can be reversed if the gene gets turned on, even in adulthood (right).
The Simons Foundation Autism Research Initiative (SFARI) currently funds more than 350 researchers, known as SFARI Investigators, who carry out groundbreaking autism research across the globe. Their work spans a wide range of subjects — from building brain cells from scratch in the lab to understanding how hypersensitivity to touch may relate to social difficulties. Below are some highlights of SFARI Investigators’ research in 2016.
Reversing Autism Symptoms
A new study suggests that it may be possible to reverse some behavioral traits linked to a particular form of autism as late as adulthood, at least in mice. SFARI Investigator Guoping Feng of the Massachusetts Institute of Technology and his collaborators looked at mice with nonfunctioning copies of SHANK3, an autism risk gene believed to contribute to about 1 percent of all autism cases. They found that these mice can recover from traits such as obsessive grooming and social deficits if the gene gets turned on, even in adulthood.
SHANK3 is one of many genes involved in the formation of synapses, the junctions between neurons that allow them to send signals to each other. In mice lacking SHANK3, neurons in a brain region called the striatum communicate at synapses less effectively than do those of control mice. But turning the gene on in adulthood reverses this deficit and also rescues the mice’s obsessive grooming and social deficits, the researchers reported February 25, 2016, in Nature.
Activating SHANK3 in the adult mice does not reverse all the traits related to the mutation: The mice continue to show signs of anxiety and motor difficulties, such as having trouble balancing on a rotating rod. But when the researchers activated SHANK3 in three-week-old mice — an age corresponding to childhood in people — the mice’s motor deficits eased and, to some extent, so did their anxiety. The findings highlight the importance of identifying developmental windows in which autism therapies may be administered with the greatest efficacy. Yet they also offer hope that some symptoms may be reversible even late in life.
Touching Anxiety
Hypersensitivity to touch may be largely responsible for the anxiety and social difficulties that are hallmarks of autism, a new mouse study suggests. Mice engineered to lack one of two autism-linked genes (MECP2 or GABRB3) in neurons that relay touch signals to the brain and spinal cord develop anxiety and social problems later in life. SFARI Investigator David Ginty of Harvard Medical School and his colleagues reported the findings July 14, 2016, in Cell. By contrast, mice that lack MECP2 in some cortical neurons but not in the touch neurons outside the brain show no differences in anxiety or social behavior compared with healthy mice.
Video Credits: Ginty Lab/Howard Hughes Medical Institute
Mice with a deletion of the MeCP2 gene in peripheral somatosensory neurons exhibit more anxiety-like behavior than their siblings. In this experiment, Ginty and colleagues placed mice in an unfamiliar environment. The mouse with the MeCP2 deletion (right) spends much more time closer to the walls of the chamber and less time moving around the chamber in general compared with a mouse without the deletion (left). Black lines track the movements of the mice over time and the videos are played back at 10 times actual speed.
Ginty’s team found that mice lacking two other autism-related genes, SHANK3 and FMR1, are also hypersensitive to touch stimuli. And in mice lacking MECP2 or GABRB3, touch signals run to the spinal cord without the dampening that usually happens in healthy mice. Many people with autism report heightened sensitivity to touch, and the new results support the idea that this sensitivity may underlie these individuals’ social difficulties.
Although autism researchers tend to focus on the brain, the new study suggests that treatments that target the peripheral nervous system — which connects limbs and organs to the brain or spinal cord — may play an important role in easing the social problems associated with autism. Unlike drugs that act on brain cells, drugs that act on peripheral neurons do not have to cross the blood-brain barrier, which greatly expands the range of potential therapies. Such treatments may need to be administered early in development, however: Ginty and his colleagues found that turning off the genes in peripheral neurons affects social behavior only if done before adulthood.
Unpacking the Mechanisms of an Autism Disorder
A mouse study published October 19, 2016, in Neuron pinpoints some of the biological underpinnings of Smith-Magenis syndrome, a rare condition linked to autism. The syndrome, caused by a deficiency of a gene called RAI1, is characterized by motor problems, obesity, intellectual disability and social difficulties. Similarly, mice that lack RAI1 are obese and struggle with learning, memory and balance.
Researchers had previously established that RAI1 controls the expression of other genes, but they didn’t know which genes. The new study, led by SFARI Investigator Liqun Luo of Stanford University, has identified a subgroup of these genes that are involved in the assembly of neuronal circuits. By examining mice that lack RAI1 in particular types of brain cells, the research team has provided insight into how the gene’s absence produces the symptoms of Smith-Magenis syndrome.
The researchers found that the loss of RAI1 in excitatory neurons (ones that promote brain activity) has no apparent effect when the gene is deleted in the cerebral cortex. But if the gene is absent in other parts of the brain, the mice develop motor problems, intellectual deficiencies and obesity. In particular, deleting RAI1 from excitatory neurons in the hypothalamus dampens expression of HTR2C and BDNF, two genes involved in reducing appetite. The study suggests that a drug that activates HTR2C, already approved by the U.S. Food and Drug Administration to treat obesity, may help individuals with Smith-Magenis syndrome control their weight.

Image Credit: Liqun Luo Laboratory/Stanford University
Mice deficient in the gene Rai1 are characteristically obese with learning, memory and balance problems. In normal adult mice, the gene is widely expressed through the brain (yellow marks regions with higher expression).
Pinning Down Microglia’s Role
Two recent studies by SFARI researchers have offered both a toolbox for studying microglia — the brain’s resident immune cells — and new insights into their role in Rett syndrome. This autism-related syndrome is marked by cognitive and physical regression, breathing and movement difficulties, and intellectual disability.
Microglia, which are involved not only in destroying pathogens but also in pruning synapses, are thought to play a role in a variety of neurological disorders, such as Parkinson’s disease, Alzheimer’s disease and schizophrenia. It has been difficult for researchers to assess these cells’ roles in detail, however. Until recently, the only way to study human microglia in the lab was to use microglia from postmortem brains, which are in short supply.
Now, a team led by SFARI Investigator Rudolf Jaenisch of the Massachusetts Institute of Technology has developed a method for making microglia out of stem cells reprogrammed from skin cells. The microglia the researchers created express many of the behaviors of microglia in living people — for example, they are capable of engulfing small objects, move through a culture of neurons and other brain cells, and travel quickly toward injured brain cells.
The team also grew microglia from a stem-cell line that carries a mutation in MECP2, the gene responsible for Rett syndrome. These microglia were exceptionally small, offering researchers a hint about the cells’ role in the condition.
Meanwhile, however, a second study suggests that MECP2 mutations in microglia are not the primary driver of Rett syndrome. Instead, microglia’s role may come into play largely in the final stages of the syndrome.
The research team behind the study, led by SFARI Investigator Beth Stevens of Boston Children’s Hospital, had previously shown that microglia prune weaker synapses in the brains of healthy mice as part of normal development. In the new work, published July 26, 2016 in eLife, Stevens’ group found that microglia in mice with Rett syndrome prune too many connections — but only near the end of the mouse’s life. This suggests that microglia do not play a causative role in the disorder, but instead exacerbate it.

Image Credit: D.P. Schafer et al/Elife 2016
Immune cells in the brain called microglia (green) engulf structures that connect neurons with other neurons or muscle cells (red) at higher rates in mice with MeCP2 mutations (right) than in mice without the mutation (left).
Knocking out MECP2 only in microglia does not produce Rett-like symptoms in mice, the team found, and these microglia behave normally. The study suggests that microglia contribute to the end stage of the condition by dismantling circuits weakened by the loss of MECP2 in other types of cells in the central nervous system.

