GIANT
Genome-wide Scale functional interaction networks for 144 human tissues and cell types
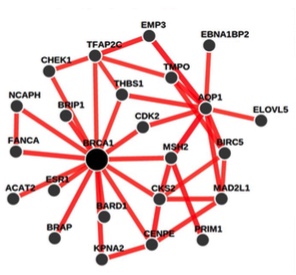
We present genome-wide functional interaction networks for 144 human tissues and cell types developed using a data-driven Bayesian methodology that integrates thousands of diverse experiments spanning tissue and disease states. Tissue-specific networks predict lineage-specific responses to perturbation, identify the changing functional roles of genes across tissues and illuminate relationships among diseases. We introduce NetWAS, which combines genes with nominally significant genome-wide association study (GWAS) P values and tissue-specific networks to identify disease-gene associations more accurately than GWAS alone. Our Web server, GIANT, provides an interface for exploring human tissue networks through multi-gene queries, network visualization, analysis tools including NetWAS, and downloadable networks. GIANT enables systematic exploration of the landscape of interacting genes that shape specialized cellular functions across more than a hundred human tissues and cell types.
IMP 2.0
Integrative Multi-Species Prediction
IMP, originally released in 2012, is an interactive Web server that enables molecular biologists to interpret experimental results and to generate hypotheses in the context of a large cross-organism compendium of functional predictions and networks. The system provides biologists with a framework in which to analyze their candidate gene sets in the context of functional networks, expanding or refining their sets using functional relationships predicted from integrated high-throughput data. IMP 2.0 integrates updated prior knowledge and data collections from the last three years in the seven supported organisms (Homo sapiens, Mus musculus, Rattus norvegicus, Drosophila melanogaster, Danio rerio, Caenorhabditis elegans, and Saccharomyces cerevisiae) and extends function prediction coverage to include human disease. IMP identifies homologs with conserved functional roles for disease knowledge transfer, allowing biologists to analyze disease contexts and predictions across all organisms. Additionally, IMP 2.0 implements a new flexible platform for experts to generate custom hypotheses about biological processes or diseases, making sophisticated data-driven methods easily accessible to researchers.
FNTM
Functional Networks of Tissues in Mouse

FNTM provides biomedical researchers with tissue-specific predictions of functional relationships between proteins in the most widely used model organism for human disease, the laboratory mouse. Users can explore FNTM-predicted functional relationships for their tissues and genes of interest or examine gene function and interaction predictions across multiple tissues, all through an interactive, multi-tissue network browser. FNTM makes predictions based on integration of a variety of functional genomic data, including over 13,000 gene expression experiments, and prior knowledge of gene function. FNTM is an ideal starting point for clinical and translational researchers considering a mouse model for their disease of interest, for researchers already working with mouse models who are interested in discovering new genes related to their pathways or phenotypes of interest, and for biologists working with other organisms to explore the functional relationships of their genes of interest in specific mouse tissue contexts. FNTM predicts tissue-specific functional relationships in 200 tissues, does not require any registration or installation, and is freely available for use at http://fntm.princeton.edu.
DeepSEA
Deep learning-based algorithmic framework for predicting chromatin effects
DeepSEA is a deep learning-based algorithmic framework for predicting the chromatin effects of sequence alterations with single-nucleotide sensitivity. DeepSEA can accurately predict the epigenetic state of a sequence, including transcription-factor binding, DNase I sensitivities, and histone marks in multiple cell types, and it can further utilize this capability to predict the chromatin effects of sequence variants and prioritize regulatory variants.
SEEK
Search-based exploration of expression compendium

SEEK is a query-based search engine for very large transcriptomic data collections, including thousands of human datasets from many different microarray and high-throughput sequencing platforms. SEEK uses a query-level cross-validation-based algorithm to automatically prioritize datasets relevant to the query and takes a robust search approach to identify genes, pathways and processes coregulated with the query. SEEK provides multigene query searching with iterative metadata-based search refinement and extensive visualization-based analysis options.
URSA
A data-driven perspective your gene expression assay

URSA (Unveiling RNA Sample Annotation) leverages the complex tissue/cell-type relationships and simultaneously estimates the probabilities associated with hundreds of tissues and cell-types for any given gene-expression profile. URSA provides accurate and intuitive probability values for expression profiles across independent studies and outperforms other methods irrespective of data preprocessing techniques. Moreover, without re-training, URSA can be used to classify samples from diverse microarray platforms and even from next-generation sequencing technology.
Nano-Dissection
In silico nano-dissection
Cell-lineage-specific transcripts are essential for differentiated tissue function in metazoan organisms. These transcripts are frequently found to be the cause of hereditary disease, and they mediate progression of acquired diseases. Identifying the tissue-specific transcriptome can guide disease gene identification in genetic studies and the development of organ specific therapeutic targets. This server performs an in silico nano-dissection, an approach we developed to identify genes with novel cell-lineage specific expression. This bioinformatics strategy leverages high-throughput functional genomics data from tissue homogenates to accurately predict genes enriched in specific cell types.
PILGRM
PILGRM is for the biologist with a set of proteins relevant to a disease, biological function or tissue of interest who wants to find additional players in that process. It uses a data-driven method that provides added value for literature search results by mining compendia of publicly available gene-expression datasets using lists of relevant and irrelevant genes (standards).
HEFalMp
Human Experimental/Functional Mapper
A functional map is a way of usefully exploring information from thousands of experimental results, focusing on a specific query of interest. This might mean finding data that pertain to a single gene/protein, a group of related (or unrelated) genes, a pathway, a process, or a set of disease-related genes. Functional maps rely on data integration to summarize genomic data as functional relationship networks. Each network encodes how likely it is for every pair of genes in the genome to interact functionally — possibly through a direct interaction, like protein binding, or through an indirect functional relationship, such as participation in the same cellular process. Functional mapping analyzes portions of these networks that relate to user-specified groups of genes and biological processes and displays the results as probabilities (for individual genes), as functional association P-values (for groups of genes), or graphically (as an interaction network). HEFalMp contains information from roughly 15,000 microarray conditions, over 15,000 publications on genetic and physical protein interactions, and several types of DNA and protein sequence analyses, and it allows exploration of over 200 process-specific functional relationship networks in humans, including a global, process-independent network that captures the most general functional relationships.
Sleipnir
Sleipnir Library for Computational Functional Genomics

Sleipnir is a C++ library that enables efficient analysis, integration, mining and machine learning involving genomic data. This includes a particular focus on microarrays, since they make up the bulk of available data for many organisms, but Sleipnir can also integrate a wide variety of other data types, from pairwise physical interactions to sequence similarity or shared transcription-factor binding sites. All analysis is done with attention to speed and memory usage, enabling the integration of hundreds of datasets covering tens of thousands of genes. In addition to the core library, Sleipnir comes with a variety of pre-made tools, providing solutions to common data-processing tasks and examples to help researchers use Sleipnir in their own programs. Sleipnir is free, open-source, fully documented and ready to be used by itself or as a component in computational biology analyses.
KNNimpute
K-Nearest Neighbors Imputation

KNNimpute is an implementation of the k-nearest neighbors algorithm for estimation of missing values in microarray data. In our comparative study of several different methods used for missing-value estimation, we determined that KNNimpute provides superior performance in a variety of situations.