
Harnessing the Power of Evolution in the Battle Against COVID-19
Many people are surprised to learn that their chances of surviving an infectious disease rest largely in their genes. Differences in DNA can give some people an advantage over others, protecting them from infection or from its most devastating effects. But infectious pathogens can still pose a threat by mutating or evolving to evade our defenses—for example, by jumping from an animal species to humans or by developing resistance to once-effective drugs. Indeed, a battle between hosts and pathogens has been taking place for millennia. Decades of basic science research have helped us to understand this evolutionary arms race and stay one step ahead of the pathogens in our midst—by informing the development of cutting-edge therapeutics and vaccines to combat the COVID-19 pandemic and by preparing us for the outbreaks still to come.
An enigma emerges
In the 1990s, while the world was dealing with a frightening new virus, HIV, that had grown to be the fourth-biggest killer globally, scientists noticed a peculiar trend. There were a few lucky people who, despite being exposed repeatedly to HIV, the virus that causes AIDS, never became infected. Scientists raced to find the secret to this unusual immunity. In the summer of 1996, they discovered that people who possess two mutant copies of the gene for CCR5—the protein that HIV uses to infect immune cells—were protected against the virus.
These findings spawned theories that the protective mutations might be an evolutionary relic of some ancient viral disease, retained in the genomes of the descendants of people who had survived diseases like the black plague, smallpox, or dysentery. However, after geneticists detected the CCR5 mutation in the skeletal remains of Bronze Age humans, at a frequency similar to that of modern-day people, those theories were put to rest. Still, this research served as a reminder that the human genome can shape the course of an infectious disease and that our encounters with infectious disease can shape us.
The discovery of CCR5 was a significant waypoint in the hunt for a potential cure for HIV. Who knows what advances will come from studying the genetics of SARS-CoV-2, the new coronavirus that causes COVID-19?
Long before the discovery of the CCR5 mutation, scientists had begun to suspect that our genes play a role in a person’s vulnerability to infection. By the turn of the 20th century, basic science research studies were already showing that most infections in plants, animals, and humans were actually benign or silent. Although germ theory had earlier established that microorganisms are the cause of dreaded communicable diseases, these newer studies indicated that only a small proportion of all people infected with a given microbe get sick. How was that possible?
“That’s what we call the infection enigma,” says Jean-Laurent Casanova, MD, PhD, a geneticist at Rockefeller University.
Over time, scientists started to wonder whether humans were actually evolving to develop resistance to infectious diseases. In the 1950s, they discovered that a genetic blood disorder known as sickle-cell disease was more common in Africa where it helps protect people from the mosquito-borne illness malaria. This finding provided one of the first pieces of molecular evidence that natural selection operates in humans, as the mutations causing sickle-cell disease appear more frequently than could be explained by chance. In other cases, mutations do not confer an advantage but still occur repeatedly in the human population. These genes might never become common, but they can have devastating effects. Striking examples emerged when scientists discovered the genetic mutations responsible for what are known as primary immunodeficiencies—disorders that result in multiple, recurrent infections so severe that those affected often do not live past childhood.
When Casanova started his lab in 1996, he was already convinced that similar, albeit more subtle genetic defects, which he calls inborn errors of immunity, were lurking in the general population. He thought that rather than making a person susceptible to all infections, these defects might be a chink in the person’s defensive armor, leaving them vulnerable to a specific pathogen.
“As long as a person doesn’t encounter that pathogen, the mutation will go unnoticed,” says Casanova, who is today funded as a Howard Hughes Medical Institute (HHMI) Investigator.
Born and educated in France, Casanova has spent years amassing blood samples from previously healthy patients who experience surprisingly severe infections, then combing through their genomes to look for tiny changes that might explain why they fell ill. For some infections, he has discovered a single defect in one patient. For others, like influenza, herpes simplex virus, and tuberculosis, he has identified defects that explain a significant proportion of cases. For example, one in every three people are infected with the tuberculosis-causing bacteria Mycobacterium tuberculosis. Most, however, do not show signs of the disease. But Casanova and his team learned that people with two mutant copies of a gene called TYK2 are more likely to develop the illness. According to Casanova, such errors in the genome are as necessary as the microbes themselves for the development of infectious diseases.
Other scientists agree. Priya Duggal, PhD, a genetic epidemiologist at Johns Hopkins, was in graduate school when the CCR5 mutation was identified. She remembers being drawn to the idea that the human genome could affect an individual’s risk of infection. “We knew a lot at that point about how the human genome played a role in cancer and cardiovascular disease,” she says, “but until then, I had never thought about its role in infectious diseases.” During her postdoctoral fellowship at the National Human Genome Research Institute, Duggal says, people warned her that focusing on infectious diseases might be a bad career move. “They told me I should just study cancer, because it was a recognized problem and one we could try to address with genetics.”
Since starting a lab of her own, Duggal has uncovered a genetic basis for vulnerability to several infections, including HIV, Entamoeba histolytica, and hepatitis C. For example, she has shown that variation in the human leukocyte antigen (HLA) genes, which influence the body’s immune response, may explain why some people are able to spontaneously get rid of hepatitis C infection whereas others are left with chronic disease.
It’s in our genes
With all of the groundwork laid by geneticists like Casanova and Duggal, it should have come as no surprise that COVID-19 would hit some people harder than others. Yet the striking difference in outcomes from infection with SARS-CoV-2 remains one of the most perplexing aspects of this 21st-century pandemic. Scientists estimate that approximately half of those infected never even feel sick, whereas others end up in the hospital fighting for their lives. Although the elderly, those with preexisting conditions, and men are more likely to suffer severe complications, thousands of young and previously healthy people who were infected in 2020 still haven’t recovered fully from the disease.
Contrary to what some people may think, Casanova says there is nothing random about who gets sick. “It goes back to this concept of biological determinism, which means that there are causes and there are consequences,” he says. “If you’re in the ICU, there’s a reason, and it’s not because you had an argument with your mother-in-law or you had Brussels sprouts for breakfast.”
Rather, Casanova contends, it’s likely due to genetics.
Soon after Casanova heard about the then-new respiratory virus in China, he launched the COVID Human Genetic Effort, an international consortium of more than 50 sequencing hubs and hundreds of hospitals aimed at discovering inborn errors of immunity relevant to COVID-19. As he had done in his studies of other illnesses, Casanova collected blood samples from thousands of hospitalized patients and searched their genes for telltale defects. Several months into the effort, Casanova’s team made two major advances. They found that 4% of people with life-threatening cases of COVID-19 carry mutations affecting type-1 interferons, proteins that typically protect against viruses. Another 10% of patients had rogue autoantibodies—that is, antibodies that attack not a virus but a component of the host’s immune system, specifically those same type-1 interferons. Defects in this part of the immune response, his research suggests, may be responsible for some of the most severe cases of COVID-19.
“It is mind-blowing,” says Casanova, who calls the discovery the most exciting of his career. “These people were walking down the street with these antibodies and were fine, then they get hit by SARS-CoV-2 and a week later they are in the ICU.”
More insights into the COVID-19 infection enigma have emerged from the research of other teams around the globe. One study suggests that variants in the HLA genes, which Duggal studies, likely play a role. Others point to differences in an individual’s blood type, as well as to variants in the ACE2 gene, which codes for the protein on human cells that SARS-CoV-2 latches onto. For her part, Duggal is leading a study comparing the DNA of patients hospitalized at Johns Hopkins with that of people in the community who tested positive but were never hospitalized. She has been surprised to find that many of the study participants are so-called “long-haulers,” who experience persistent symptoms months after being infected.
“Genetics is a small portion of this big story,” says Duggal, adding that socioeconomic and structural factors also can put certain populations at higher risk. “I am the first to acknowledge that there’s a lot more going on that we don’t understand.”
Signatures of evolution
When it comes to the evolutionary arms race between humans and viruses, viruses appear to have the upper hand. Viruses exist in a nether world between the living and the nonliving, their only task being to insert their genetic material into a host—whether a bat, a human, or some other creature—which then unwittingly copies the viral genome along with its own. Because viruses are so small, their genetic material can be replicated quickly: the genome of SARS-CoV-2 contains 30,000 “letters,” compared with a human’s 3 billion. And because they typically incorporate typos into their genome at a high rate, viruses can quickly adapt as conditions change.
However, the fact that viruses mutate is not necessarily cause for alarm. That’s because most mutations are as bad for the virus as they are for its host, says Jesse Bloom, PhD, an evolutionary biologist at Seattle’s Fred Hutchison Cancer Research Center.
“It’s not like if you took a human and dialed up their mutation rate a millionfold they would evolve the ability to perform superhuman feats,” Bloom says. “They would probably end up dead, and the same is true for viruses.” In other words, a genetic change must bestow an advantage and undergo natural selection—as was the case with Darwin’s finches—before a species can evolve into a fitter or more formidable version of itself.
Some viruses evolve more quickly than others. Most RNA viruses like HIV and Ebola lack the “proofreading” capability of DNA viruses and are thus more prone to errors that allow especially fast evolution. The pressure to adapt is also stronger in viruses that cause chronic diseases, as the viral machinery has to constantly contend with the host’s immune system. The notorious shape-shifting ability of the flu virus, for example, which can evolve up to a million times faster than humans do, explains why we have to get a new flu shot every year.
Even so, Bloom says, it can take years for the flu to evolve to the point that it can escape our immune defenses and reinfect a previous flu victim. Bloom uses a sophisticated technique called deep mutational scanning to study this evolution in the lab. His team makes tens of thousands of tweaks to viruses and then measures how the alterations affect their function. For example, the researchers tested how mutations to the flu virus affected its ability to be recognized by antibodies from 16 study volunteers. They were surprised to find that though the human immune system can potentially make an astronomical number of antibodies, the actual immune response in many people is narrowly focused on a specific part of the virus.
“I think that’s incredible,” says Bloom, whose team has also used this technique to study HIV, Zika, and SARS-CoV-2. “We found that different people’s immunity is focused in different ways, meaning that they may be more protected or more susceptible to whatever strain is circulating. It also means that as the virus is evolving, there may be new mutations that are strongly favored while the virus is replicating in one person, but that are no longer favored when the virus jumps to another person. It presents a lot of interesting evolutionary implications.”
Going viral
For most of us, it wasn’t until this new and dangerous pathogen emerged in our midst that we began to suspect the presence of viruses everywhere—potentially lurking on the handles of grocery carts, or launched by the laughter of neighborhood children, or hanging in the air like smoke in a bar. In reality, an estimated 1 × 1031 (a quadrillion quadrillion) individual viruses exist in the world. But most of these viruses do not make us sick. The precursor to SARS-CoV-2 likely circulated in horseshoe bats for millennia before mutating into a version that could jump—perhaps through an intermediate species like the scaly anteater—to humans. Since then, it has continued to evolve, though recent changes have been far less dramatic, says Emma Hodcroft, PhD, a quantitative geneticist at the University of Bern in Switzerland.
The British-American researcher is a codeveloper of Nextstrain.org, an online tool that tracks the evolution of pathogens in real time. She says that while most viral mutations do not affect the behavior of the virus, they do act like molecular “breadcrumbs” that scientists can use to retrace the virus’s steps as it moves through the human population. The Nextstrain platform compiles mutation data from all over the world and uses it to construct what are essentially family trees for viruses. These colorful, branching “phylogenetic” trees had already generated key insights into the outbreaks of Ebola in 2014 and Zika in 2015, and into the longer-term spread of influenza, before they were applied to SARS-CoV-2.
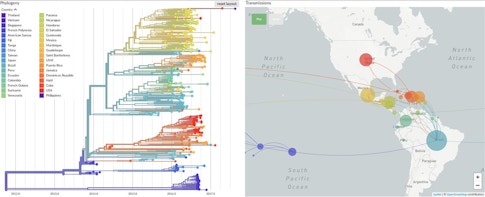
In the first year of the COVID-19 pandemic, scientists sequenced more than 420,000 SARS-CoV-2 genomes, each one a variant of the original virus that first infected humans. Hodcroft says that this new coronavirus is mutating slowly and that most of the mutations her team has seen so far are harmless. One variant, called D614G, emerged early in the pandemic and has since all but replaced other variants globally. Research has suggested that this mutation made the virus more contagious, but Hodcroft says other factors could also cause a particular variant to dominate.
“It could just be in the right place at the right time,” she says, “at the right superspreader event, to start expanding in a population.” Take a well-studied coronavirus superspreader event in the U.S., a conference organized by the biotech company Biogen at a Boston hotel in February 2020. Initial reports found that 99 people became infected at the event. But a later phylogenetic analysis by dozens of researchers, including Harvard computational geneticist Pardis Sabeti, MD, PhD, showed that the coronavirus strain unleashed at the meeting went on to spawn 300,000 more cases by October. The team’s genetic sleuthing was aided by the discovery that the infected conference-goers carried a strain of coronavirus known as C2416T, which at the time had been seen only in France. They tracked C2416T as it went on to spread through 29 states and three countries.
In December 2020, virus trackers discovered a new variant called B.1.1.7, which had emerged in the U.K. with an unusually large number of mutations. Several studies showed that unlike many of its predecessors, this variant did behave differently, spreading more quickly and easily through the population. The discovery raised global alarms as people feared that the new variant also carried mutations that could render useless the vaccines then just emerging from the approval pipeline. In a Twitter thread detailing his research (now published in a peer-reviewed journal), Bloom stated that he was confident that early vaccines will remain useful for a while, even if evolution starts to erode their effectiveness. He has used his deep mutational scanning technique to measure how mutations in the coronavirus’s spike proteins, which adorn its surface like a crown, help SARS-CoV-2 evade the immune system. His team found that one mutation, called E484K, eroded the ability of antibodies from recovered COVID-19 patients to block the virus. That mutation was not present in the U.K. variant but has popped up in another variant that emerged in South Africa. Still, Bloom says that it would take several precisely positioned mutations, likely accumulated over years of evolution, before the effectiveness of vaccines could be eliminated completely.
The upper hand
Even if the coronavirus evolves resistance to our antibodies and vaccines, Bloom and other scientists will remain hopeful. Both the Pfizer and Moderna COVID-19 vaccines—the first two to receive Emergency Use Authorization from the U.S. Food and Drug Administration—are based on mRNA, strips of genetic code that can easily be modified to accommodate any consequential new mutations in the virus. As platforms for making vaccines and antibodies become faster, our ability to understand viral evolution will be increasingly useful, because such knowledge will enable researchers to actually respond in real time while the virus is evolving.
The COVID-19 pandemic is the fifth major outbreak that Sabeti has investigated firsthand. “Through each outbreak, I have found that every time one subsides, things go back to normal and funding becomes even harder to find,” she says. “COVID-19 clearly has been a transformative event in that it has touched everyone, now that the entire world is engaged and aware of it. This time around, I cannot imagine that things will go back to normal in the way that they have before. I am cautiously hopeful that we will finally recognize that we need to invest in basic science research.” (Funders in her own research, as well as Bloom’s, include HHMI and the Burroughs Wellcome Fund.)
Science has shown us that viruses are a fundamental part of ourselves and of the world. Millions of us are infected with pathogens that we may never know we carry. Previous infiltrations have left our DNA riddled with bits of viral code, which now make up to half of the human genome. In a few cases, humans have even made adjustments to our own DNA—for example, by engineering the immunity conferred by the CCR5 mutation that was discovered a quarter of a century ago.
And scientists now have more weapons in their arsenal than ever before. Indeed, though the evolutionary arms race rages on, investments in basic science research may finally have given humans an advantage.
About the Author
Marla Broadfoot is a science journalist with a Ph.D. in genetics and molecular biology. Her work has appeared in Scientific American, STAT, Science, and Nature News, among others.
This article first appeared as part of the Science Philanthropy Alliance‘s COVID-19 Basic Science Prequels series. Decades before COVID-19 gripped the world, basic science researchers were laying the groundwork for lifesaving medical and technological breakthroughs now central to the world’s pandemic response. With support from the Kavli Foundation and the Simons Foundation, the Science Philanthropy Alliance has enlisted a team of science writers to explore these science origin stories. Each month, the COVID-19 Basic Science Prequels will unpack the people, history, and serendipitous discovery behind topics that now dominate our daily lives.